Hernández-Frias, A. et al. RSCTyH, Vol. 1, Num.01, Enero 2023
Resumen: El síndrome de Gilbert se caracteriza por hiperbilirrubinemia no conjugada crónica, en ausencia de enfermedad hepática o hemolítica. Es una alteración hereditaria causada por un polimorfismo en la región promotora de la UDP-glucoronosiltransferasa 1 (UGT1A1) provocando la inserción de 2 bases nitrogenadas en la región promotora A(TA)6TAA de lo que resulta una elongación en la caja TATA [A(TA)7TAA] que se refleja en una expresión reducida de la enzima UGT1A1. Otros genotipos han sido descritos para dicha región promotora: TA5, TA6, TA7, TA8, pudiéndose encontrar en estado homocigoto y combinaciones de los alelos en estado heterocigoto. El presente estudio tiene la finalidad de estimar la frecuencia del síndrome de Gilbert mediante el análisis del promotor del gen UGT1A1 en individuos originarios del Estado de Sinaloa. Se recolectaron un total de 388 muestras de donadores pertenecientes a los municipios de Ahome (n=105), Culiacán (n=109), Guasave (n=65) y Mazatlán (n=109). Los genotipos identificados en la población sinaloense fueron: (TA)6/(TA)6, (TA)6/(TA)7 y (TA)7/(TA)7, con porcentajes de 44, 47 y 9% respectivamente. El síndrome de Gilbert está presente en el 9% de la población sinaloense. Los genotipos observados en nuestra investigación son los mismos que se identificaron en población asiática y europea, variando de estas su frecuencia. Con estos resultados podemos inferir que la población sinaloense tiene un fuerte componente europeo, aunque también esos genotipos fueros identificados en asiáticos; las frecuencias genotípicas obtenidas son similares a la de los primeros. Esta investigación aporta evidencia científica de hechos basados por recopilación histórica. Palabras clave: Polimorfismos, Sinaloenses, Síndrome de Gilbert |
Abstract: Gilbert’s syndrome is characterized by non-conjugated chronic hyperbilirubinemia, in the absence of hemolytic or hepatic disease. An inherited alteration caused by a polymorphism in the UDP glucuronosyltransferase 1 family, polypeptide A1 gene (UGT1A1) with an insertion of two base pairs in its promoter region A(TA)7TAA, producing a reduced UGT1A1 enzyme expression. Other genotypes have been described for this region: TA5, TA6, TA7, and TA8, found in a homozygous state and heterozygous through the combination of the alleles. The present study aimed to estimate Gilbert’s syndrome frequency in Sinaloa state native individuals through the UGT1A1 gene promoter analyses. A total of 388 samples were collected from the municipals of Ahome (n = 105), Culiacán (n = 109), Guasave (n = 65), and Mazatlán (n = 109). The genotypes identified in the Sinaloa’s population were: (TA)6/(TA)6, (TA)6/(TA)7, and (TA)7/(TA)7, with a respective percentage of 44, 47, and 9 respectably; indicating that Gilbert’s syndrome is present in 9% of the Sinaloa’s population. Gilbert’s syndrome is present in 9% of Sinaloa’s individuals. The genotypes identified in this study were similar to those found in the Asian and European populations, with a slight variation in their frequencies. Such findings allow us to infer that Sinaloa’s population has a strong European component; although these genotypes were also identified in Asians the frequencies are more similar to the first ones. This research provides scientific evidence of historically compiled facts. Keywords: Gilbert’s syndrome, Polymorphisms, Sinaloa |
Recibido: XXXXXXX; Aceptado: XXXXXX; Publicado:
Introducción
El síndrome de Gilbert pertenece al grupo de desórdenes metabólicos más comunes, se estima que del 6 al 9% de población general a nivel mundial se encuentra afectada por este síndrome [1].
Se caracteriza por la existencia de hiperbilirrubinemia no conjugada crónica, que aparecen en ausencia de enfermedad hepática o hemolítica, es una alteración hereditaria y asintomática [2], [3].
Se han reportado diferencias nucleotídicas en la región TATAA del promotor. La inserción de dos bases nitrogenadas (TA) en la región promotora A(TA)6TAA da como resultado una elongación A(TA)7TAA en la caja TATA, interfiriendo con el enlace en el factor de transcripción IID y se observa una expresión reducida de la enzima UGT1A1. En estudios realizados anteriormente se demostró que un polimorfismo en el promotor de la enzima causa el síndrome de Gilbert, una forma benigna de bilirrubinemia no conjugada. Los promotores que contienen A(TA)7TAA resultaron ser menos activos que los de tipo silvestre A(TA)6TAA [2], [4], [5].
Estudios previos han identificado mutaciones adicionales, por ejemplo, en algunos pacientes de Asia con el síndrome de Gilbert, no se encontraron mutaciones en la región promotora, pero si se encontró que son heterocigotos para las mutaciones de cambio de sentido Gly71Arg en la región codificante [6]–[9]
Se han descrito genotipos diferentes con respecto a la región promotora del gen UGTA1 (TA5, TA6, TA7, TA8) los cuales se presentan en estado homocigoto o heterocigoto. Se creía que los genotipos TA5 y TA8 eran exclusivos de individuos africanos, pero en estudios realizados por Arámbula y Vaca en 2001, se demostró la presencia de estos polimorfismos en mestizos mexicanos, lo cual podría ser causado por las mezclas de razas [10], [11].
El gen que codifica para la enzima UDP glucuronosiltransferasa UGT1A1 se localiza en el cromosoma 2 en la región 2q37. La enzima UGTisoforma 1A1 es codificada por el exón 1A1 y los exones comunes 2-5del complejo de genes. El gen de la UGT1A1 está localizado en un locus complejo que codifica varias UDP-glucuronosil transferasas [2], [12] El gen de la UGT1A1 controla la conjugación de bilirrubina ya que es el gen responsable de determinar la estructura de la enzima glucuronosiltransferasa, que es sintetizada en el hepatocito. Una región adicional no codificante adyacente al gen es el promotor, región localizada río arriba la cual es encargada de controlar la expresión genética [2], [12].
La variación de nucleótidos en la región promotora se traducirá en una enzima de estructura normal, pero con una expresión disminuida. Las mutaciones en el exón 1A1 o su promotor puede producir deficiencias estructurales o funcionales de la enzima que pueden resultar en la conjugación de bilirrubina disminuida y como consecuencia, hiperbilirrubinemia. Ejemplos de estas mutaciones deletéreas se encuentran en el síndrome de Gilbert y el síndrome Crigler-Najjar (C-N) [2], [12], [13].
En Sinaloa no se cuenta con ningún tipo de análisis que nos muestre cuál es la frecuencia del síndrome de Gilbert, por lo que el objetivo de este estudio fue estimar la frecuencia del síndrome de Gilbert mediante el análisis del promotor del gen UGT1A1 y conocer la distribución genotípica de los alelos relacionados con este síndrome (TA5, TA6, TA7 y TA8) en individuos sanos originarios del estado de Sinaloa.
Materiales y Métodos
Muestras
El estudio realizado fue prospectivo, transversal y descriptivo. Se recolectaron un total de 388 muestras de sangre con anticoagulante EDTA de donadores de sangre voluntarios nacidos en Sinaloa e hijos de padres Sinaloenses pertenecientes a los municipios de Ahome (n= 105), Culiacán (n= 109), Guasave (n= 65) y Mazatlán (n= 109). Las muestras de sangre fueron tomadas de los bancos de sangre del Instituto Mexicano de Seguro Social de los municipios. Basados en la prevalencia estimada del síndrome de Gilbert descrita en México (10%), el tamaño de muestra de la población permite una estimación fiable de la prevalencia del Síndrome de Gilbert en Sinaloa.
PCR- PCCS
Se realizó la extracción de ADN de todos los individuos utilizando el método de Gustincich [14]. La región relevante fue amplificada por la reacción en cadena de la polimerasa bajo condiciones estándar utilizando los iniciadores 5´-CTTGGTGTATCGATTGGTTTTTG-3; 5´-TTTGCTCCTGCCAGAGGTTCG-3´ en un termociclador PXE 0.2. Todos los productos de PCR fueron analizados en geles de agarosa al 2.0%. La genotipificación se realizó con Polimorfismo Conformacional de Cadena Sencilla (PCCS), los productos de PCR fueron diluidos 1:4 en buffer de carga (formamida 95%, EDTA 20mM, Azul de bromofenoal 0.05% y Xilene Cianol al 0.05%), se calentaron a 95ºC por 5 minutos e inmediatamente puestos en hielo. 20µL de muestra fueron cargados en geles de poliacrilamida (Acrilamida al 37.9%: Bisacrilamida 1%) al 20%, TBE 1.5X, Glicerol 5% y sujetos a electroforesis a 300V por 2:30, utilizando una cámara de electroforesis vertical con buffer de corrida TBE 1.5X, la temperatura fue mantenida a 10ºC por la circulación de agua fría. Los geles fueron teñidos con nitrato de plata bajo las siguientes condiciones: 10 min de solución fijadora (etanol 10%: ácido acético 0.5%), 30 min en solución de tinción (AgNO3 al 0.3 % en solución fijadora) y 15 min en solución de revelado. El patrón de bandas obtenido fue comparado con el resultante de muestras positivas para cada los genotipos (TA)5/(TA)6, (TA)6/(TA)6, (TA)6/(TA)7 y (TA)7/(TA)7, los cuales fueron identificados previamente por secuenciación.
Resultados y Discusión
Resultados
Se recolectaron un total de 388 muestras pertenecientes a los municipios de Ahome (27%), Culiacán (28%), Guasave (17%) y Mazatlán (28%. La distribución por sexos de las muestras revela que el 95% pertenece al sexo masculino. Las distribuciones de las edades de los individuos del estudio comprendieron de los 18 hasta los 58 años, con una media de 30 y 31 años.
Análisis molecular
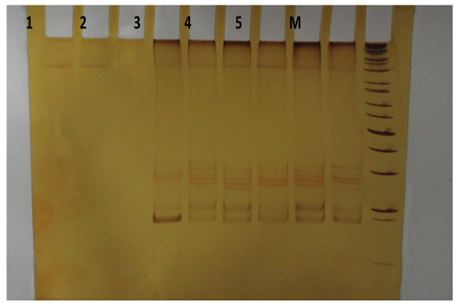
Se llevó a cabo la extracción de ADN de las 388 muestras captadas para el estudio. La integridad se verificó por electroforesis en geles de agarosa al 1.5% teñidos con gel red. Se amplificó la región de la caja TATA del gen UGT1A1 en las muestras analizadas. Los productos de PCR provenientes de la amplificación se verificaron en geles de agarosa al 2%. Los productos de PCR obtenidos de las muestras se sometieron a PCCS para identificar los polimorfismos correspondientes. Esto se realizó comparando los patrones de bandas generadas en muestras previamente genotipificadas por secuenciación (Figura1).
Figura 1. Movilidad electroforética de genotipos UGT1A1 de controles identificador por secuenciación.
Carril 1: muestra problema; carril 2: control genotipo 5/6; carril 3: control genotipo 6/6; carril 4: control genotipo 7/6; carril 5: control genotipo 7/7. M: marcador de peso molecular. Fuente: Elaboración propia
Los genotipos identificados en la población sinaloense fueron: (TA)6/(TA)6, (TA)6/(TA)7 y (TA)7/(TA)7, con porcentajes de 44, 47 y 9% respectivamente (Figura 2). Lo cual quiere decir que el 9% de los sinaloenses presentan síndrome de Gilbert.
Figura 2. Frecuencias génicas identificadas en la población sinaloense Fuente: Elaboración propia
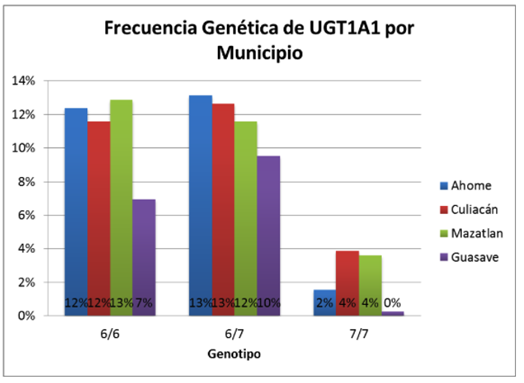
En la Figura 3 se observa la distribución de los genotipos identificados por municipio. En esta gráfica se observa que la distribución es homogénea ya que en la mayoría de los municipios se presentan porcentajes similares en los tres polimorfismos de ((TA)6/(TA)6, (TA)6/(TA)7 y (TA)7/(TA)7), solo Guasave presentó diferencias en su distribución.
Figura 3. Distribución de frecuencias genotípicas por municipios Fuente: Elaboración propia
Discusión
Se recolectaron 384 muestras de los municipios de Mazatlán (28%), Guasave (17%), Culiacán (28%) y Ahome (27%), de los cuales el 95% pertenecen al sexo masculino y el 5% restante al femenino, estas diferencias se deben a que las muestras fueron recolectadas en bancos de sangre del IMSS y la mayoría de los individuos que acuden y cumplen con los requisitos especificados por la secretaria de salud son del sexo masculino.
En análisis moleculares realizados por Beutler y col. en 1998 en distintas poblaciones a nivel mundial, se identificaron 8 genotipos diferentes. En africanos se observaron los genotipos (TA)5/(TA)6, (TA)6/(TA)6, (TA)5/(TA)7, (TA)6/(TA)7, (TA)7/(TA)7, (TA)6/(TA)8, (TA)7/(TA)8 y (TA)8/(TA)8 con frecuencias de 3, 25, 5, 36, 19, 4, 6 y 2%, respectivamente. En las poblaciones europeas y asiáticas sólo se han observado 3 genotipos: el (TA)6/(TA)6, (TA)6/(TA)7 y (TA)7/(TA)7 con el 34, 55 y 11% respectivamente para europeos y 70, 28 y 2% respectivamente para asiáticos. En estudios realizados por Arámbula y col. en el 2002 en mestizos mexicanos se identificaron 5 genotipos: el 1.9% para (TA)5/(TA)6, el 41.3% para (TA)6/(TA)6, 0.3% para el (TA)5/(TA)7, el 46.4 % para el (TA)6/(TA)7 y el 10.5% con el (TA)7/(TA)7. Los genotipos observados en nuestra investigación son los mismos que se identificaron en población asiática y europea, variando éstos en su frecuencia 44% (TA6/TA6), 47% (TA6/TA7) y 9% (TA7/TA7). Con estos resultados podemos inferir que la población sinaloense tiene un fuerte componente europeo, aunque también esos genotipos fueron identificados en asiáticos; las frecuencias genotípicas obtenidas son similares a la de los primeros.
El síndrome de Gilbert se caracteriza por la presencia de hiperbilirrubinemia no conjugada crónica, la cual aparece en ausencia de enfermedad hepática o hemolítica, es un trastorno hereditario, frecuente en población sefardí y en la población Campania de Italia [3]. La prevalencia de este síndrome a nivel mundial varía en función de los criterios de diagnóstico, éstos van desde un 2 al 19% observándose en un 11% en europeos, 2% en asiáticos y 19% en africanos. Seco y col. en el 200 describieron que el síndrome de Gilbert se encuentra en el 10% de la población española [12]. Estudios realizados por Yusoff y col. en el 2005 en Malasia, mostraron una frecuencia del 25% en recién nacidos con hiperbilirrubinemia [15]. En México se estableció previamente una prevalencia aproximada del 10.5%, y en nuestra investigación se obtuvo una prevalencia del 9% para el estado de Sinaloa.
Conclusiones
El síndrome de Gilbert está presente en el 9% de la población sinaloense. Los genotipos y las frecuencias identificadas en Sinaloa son similares a las encontradas en población europea. Esta investigación aporta evidencia científica de hechos basados por recopilación histórica. En esta población no se identificaron genotipos reportados en población africana y en otros estados de la República Mexicana.
Agradecimientos
Este trabajo fue realizado en el laboratorio de Genética y Biología Molecular de la Facultad de Ciencias Químico-Biológicas, de la Universidad Autónoma de Sinaloa. Su desarrollo se realizó con fondos del Programa de Fortalecimiento y Apoyo a Proyectos de Investigación (PROFAPI).
Conflicto de Intereses
Los autores expresan que no existen conflictos de interés al redactar el manuscrito.
Referencias
[1] J. Gil y M. M. Sąsiadek, «Gilbert syndrome: the UGT1A1*28 promoter polymorphism as a biomarker of multifactorial diseases and drug metabolism», Biomark. Med., vol. 6, n.o 2, pp. 223-230, abr. 2012, doi: 10.2217/bmm.12.4.
[2] Y. Zahedpasha, M. Ahmadpour, H. A. Niaki, y E. Alaee, «Relation between Neonatal Icter and Gilbert Syndrome in Gloucose-6-Phosphate Dehydrogenase Deficient Subjects», J. Clin. Diagn. Res. JCDR, vol. 8, n.o 3, pp. 63-65, mar. 2014, doi: 10.7860/JCDR/2014/6674.4108.
[3] B. Burchell y R. Hume, «Molecular genetic basis of Gilbert’s syndrome», J. Gastroenterol. Hepatol., vol. 14, n.o 10, pp. 960-966, oct. 1999, doi: 10.1046/j.1440-1746.1999.01984.x.
[4] P. J. Bosma et al., «The genetic basis of the reduced expression of bilirubin UDP-glucuronosyltransferase 1 in Gilbert’s syndrome», N. Engl. J. Med., vol. 333, n.o 18, pp. 1171-1175, nov. 1995, doi: 10.1056/NEJM199511023331802.
[5] V. Mlakar, S. J. Mlakar, J. Marc, y B. Ostanek, «Preparation of reference material for UGT1A1 (TA)n polymorphism genotyping», Clin. Chim. Acta Int. J. Clin. Chem., vol. 435, pp. 24-28, ago. 2014, doi: 10.1016/j.cca.2014.04.018.
[6] X. Dai et al., «A genome-wide association study for serum bilirubin levels and gene-environment interaction in a Chinese population», Genet. Epidemiol., vol. 37, n.o 3, pp. 293-300, abr. 2013, doi: 10.1002/gepi.21711.
[7] G. Chen et al., «UGT1A1 is a major locus influencing bilirubin levels in African Americans», Eur. J. Hum. Genet. EJHG, vol. 20, n.o 4, pp. 463-468, abr. 2012, doi: 10.1038/ejhg.2011.206.
[8] M. Abumiya et al., «Influence of UGT1A1 6, 27, and 28 polymorphisms on nilotinib-induced hyperbilirubinemia in Japanese patients with chronic myeloid leukemia», Drug Metab. Pharmacokinet., vol. 29, n.o 6, pp. 449-454, 2014, doi: 10.2133/dmpk.DMPK-14-RG-031.
[9] C. S. Huang, G. A. Luo, M. L. Huang, S. C. Yu, y S. S. Yang, «Variations of the bilirubin uridine-diphosphoglucuronosyl transferase 1A1 gene in healthy Taiwanese», Pharmacogenetics, vol. 10, n.o 6, pp. 539-544, ago. 2000, doi: 10.1097/00008571-200008000-00007.
[10] E. Arámbula y G. Vaca, «Genotyping by"cold single-strand conformation polymorphism" of the UGT1A1 promoter polymorphism in Mexican mestizos», Blood Cells. Mol. Dis., vol. 28, n.o 1, pp. 86-90, 2002, doi: 10.1006/bcmd.2001.0481.
[11] E. Beutler, T. Gelbart, y A. Demina, «Racial variability in the UDP-glucuronosyltransferase 1 (UGT1A1) promoter: a balanced polymorphism for regulation of bilirubin metabolism?», Proc. Natl. Acad. Sci. U. S. A., vol. 95, n.o 14, pp. 8170-8174, jul. 1998, doi: 10.1073/pnas.95.14.8170.
[12] M. L. Seco et al., «[Interest in the study of genetic variants of the promoter region of the UGT1A1 gene in neonatal jaundice]», An. Esp. Pediatr., vol. 56, n.o 2, pp. 139-143, feb. 2002.
[13] M. Kaplan et al., «(TA)n UDP-glucuronosyltransferase 1A1 promoter polymorphism in Nigerian neonates», Pediatr. Res., vol. 63, n.o 1, pp. 109-111, ene. 2008, doi: 10.1203/PDR.0b013e31815b8e7e.
[14] S. Gustincich, G. Manfioletti, G. Del Sal, C. Schneider, y P. Carninci, «A fast method for high-quality genomic DNA extraction from whole human blood», BioTechniques, vol. 11, n.o 3, pp. 298-300, 302, sep. 1991.
[15] S. Yusoff et al., «Frequencies of A(TA)7TAA, G71R, and G493R mutations of the UGT1A1 gene in the Malaysian population», Biol. Neonate, vol. 89, n.o 3, pp. 171-176, 2006, doi: 10.1159/000088844.